
Sandeep Kumar Kar1, Tanmoy Ganguly2,
1Assistant Professor; 2PDT
Department of Cardiac Anesthesiology, Institute of Postgraduate Medical Education & Research, Kolkata (India)
Correspondence: Dr Sandeep Kumar Kar, Department of Cardiac Anesthesiology, Institute of Postgraduate Medical Education & Research, Kolkata (India); E-mail: sndpkar@yahoo.co.in
ABSTRACT
Osler-Weber-Rendu disease (OWRD) or Hereditary Hemorrhagic Telangiectasia (HHT) is a rare autosomal dominant disorder that causes muco-cutanesous and visceral vascular dysplasia and results in increased tendency for bleeding. Patients with HHT presenting with continuous bleeding pose a serious problem to the Anesthesiologist .Pre-existing anemia due to recurrent bleeding is common and sudden decompensation may lead to heart failure. Uncontrolled bleeding may occur from skin lesions during patient positioning and transport. Epistaxis may lead to aspiration of blood into trachea causing pulmonary edema. Intravenous access may be difficult. Sudden change in blood pressure may cause bleeding from arteriovenous malformations (AVMs) anywhere in the body, most serious of which is from cerebral AVM. Gastric distension may occur from ingested blood and may cause reflux and aspiration during induction. Any instrumentation including laryngoscopy and intubation, nasogastric tube insertion, urinary catheterization should be carried out with utmost caution as bleeding may occur from undetected lesions. Management include blood transfusion, antifibrinolytics and surgical hemostasis. Anesthesia strategy should include rapid sequence induction and controlled hypotension.
Key words: Telangiectasia, Hereditary Hemorrhagic; Osler-Rendu Disease; Osler-Weber-Rendu Syndrome; Congenital Abnormalities; Cardiovascular Abnormalities; Vascular Malformations; Arteriovenous Malformation
Citation: Kar SK, Ganguly T. Hereditary hemorrhagic telangiectasia and the anesthesiologist. Anaesth Pain & Intensive Care 2017;21(3): 387-392
Received – 5 Mar 2017; Reviewed & Accepted – 16 Mar 2017
INTRODUCTION
Osler-Weber-Rendu disease (OWRD) or Hereditary Hemorrhagic Telangiectasia (HHT) is a rare autosomal dominant disorder that causes muco-cutanesous and visceral vascular dysplasia and results in increased tendency for bleeding.1-4 Patients with HHT may present with variety of symptoms and management differs accordingly. Epistaxis is the most common symptom of HHT and mucocutaneous telangiectasia is the most common sign.5
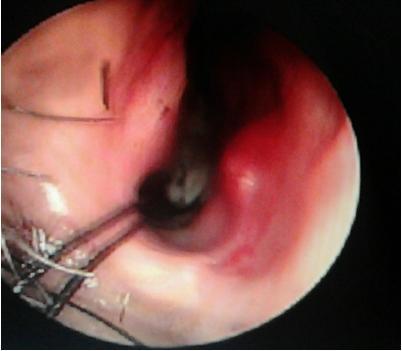
Figure 1: Endoscopic view of the angiofibroma
INCIDENCE
HHT is a rare systemic fibro vascular dysplasia6 with incidence varying from 1 in 5,000 to 10,0007 to 1 to 2 in 1,00,0006. Sutton8 in 1864 first described this syndrome in a man with a vascular malformation and recurrent epistaxis. In 1896 Rendu9 first noted the association between hereditary epistaxis and telangiectasia in a 52 years old man. Osler10 in 1901 and Weber11 in 1907 further elaborated the association between hemorrhagic lesions in skin and mucous membranes and its familial inheritance. Although the disease is popularly known as Osler-Weber-Rendu syndrome, the name ‘hereditary hemorrhagic telangiectasia’ suggested by Hanes12 in 1909, recognizes the characteristics that define the disease.
GENETICS OF HHT
HHT is manifested by mucocutaneous telangiectasias and arteriovenous malformations (AVMs) in different parts of body. Lesions can affect the nasopharynx, central nervous system (CNS), lung, liver, and spleen, as well as the urinary tract, gastrointestinal (GI) tract, conjunctiva, trunk, arms, and fingers.2,13 Impaired signaling of transforming growth factor-ß/bone morphogenesis protein (TGF-β/BMP)14-17 as well as vascular endothelial growth factor (VEGF)18,19 has been attributed as the primary cause of HHT. The gene mutations found to be responsible are as follows in Table 1.
Table 1: Types of HHT with genetic basis
| HHT types | Mutated genes | Gene location |
| HHT1 | ENG20 | Long arm of chromosome 921-23 |
| HHT2 | ALK1 (Activin receptor-like kinase 1), also called ACVRL124,25 | Long arm of chromosome 12 |
| hereditary benign telangiectasia (HBT), HHT3 | RASA1 26 | chromosome 5q14 |
| HHT4 | Chromosome 7p1427 | |
| HHT + Juvenile polyposis coli | SMAD4/MADH428-30 | 18q21.2 |
| HHT2 + primary pulmonary hypertension | BMPRII31,32 | 2q33 |
DIAGNOSIS
The diagnosis of HHT is made clinically on the basis of the Curaçao criteria3, established in June 1999 by the Scientific Advisory Board of the HHT Foundation International, Inc. (Table 2), and recommended by HHT Foundation International – Guidelines Working Group,33 or by identification of a causative mutation.
Table 2: Curaçao criteria
| Criterion | Description |
| Epistaxis | Spontaneous and recurrent |
| Telangiectasias | Multiple, at characteristic sites: lips, oral cavity, fingers, nose |
| Visceral lesions | GI Telangiectasia, pulmonary, hepatic, cerebral or spinal AVMs |
| Family history | A first degree relative with HHT according to these criteria |
“definite” if 3 or more criteria are present, “possible or suspected” if 2 criteria are present, and “unlikely” if 0 or 1 criterion is present.
Histopathology of HHT lesions show many layers of smooth muscle cells without elastic fibers and very frequently arterioles directly communicating with smooth muscle cells. As a result telangiectasias are very sensitive to slight trauma and friction. HHT may present in children as bleeding but usual age of presentation in adulthood.4 Male and females are equally affected.34 Classic triad of presentation include telangiectasias of the skin and mucous membranes, epistaxis, and a positive family history. Epistaxis may be present in upto 95% cases,4,35 whereas skin lesions account for 75-90% of presentations.35,36 Skin telangiectasias rarely cause bleeding4. Gastrointestinal telangiectasia may occur in 10-33% patients37 most commonly in the stomach and upper duodenum.37 Significant bleeding from gastrointestinal tract may occur in 25% patients older than 60 years and may increase with age.38 Pulmonary involvement in the form of arteriovenous malformations (AVMs) may be present in 75% HHT1 and 44% HHT2 patients.39 Patients with pulmonary involvement are at high risk of developing cerebral thrombotic and embolic events including stroke, brain abscess, or transient ischemic attacks due to right-to-left shunting.14,37 Cerebral AVMs may be present 15-20% HHT1 and 1-2% HHT2 patients,39-43 and may present with seizure, headache or intracranial haemorrhages.4,44 Hepatic AVMs may be present upto 74% cases45 but usually asymptomatic4.
MANAGEMENT
Management strategies for AVMs associated with HHT may differ with location and presentation and depicted in Table 3.
Table 3: Management strategy of HHT according to site of involvement
| Location | Lesions | Indications of management | Management |
| Nose | Telangiectasia, AVMs | Recurrent epistaxis | Sclerotherapy with sodium tetradecyl sulphate46, submucosal radiofrequency47, Bevacizumab48, Septal mucosal dermoplasty39,49, Embolization of external carotid artery branches50, |
| Skin | Telangiectasia | Pain2, cosmesis4 | Cauterization, hypertonic saline sclerotherapy, dye laser treatment51. Pulsed Nd:YAG laser52, |
| Gastro intestinal tracts | AVMs, Angio-dysplasia4 | Chronic anemia, melena | Diagnosis: Endoscopy, Angiography4
Management: Bipolar electrocoagulation4, Laser4, estrogen-progesterone therapy53, interferon α54 |
| Lungs | AVMs | Exercise intolerance, cyanosis, migraine headaches, polycythemia and clubbing
CNS events14,40 During pregnancy55 |
Feeder vessel >3 mm: Transcatheter embolisation56,
Smaller lesion: Follow up14 Antibiotic prophylaxis to prevent brain abscess |
| Diffuse pulmonary AVM | Severe hypoxia | Lung transplantation4 | |
| CNS | Cerebral and spinal AVMs | Transcatheter embolization, resection, stereotactic radiosurgery57,58 | |
| Liver | AVMs, | Life threatening portosystemic shunts | Liver transplant59-61
Bevacizumab62 |
Patients with HHT presenting with continuous bleeding pose a serious problem to the Anesthesiologist .Pre-existing anemia due to recurrent bleeding is common and sudden decompensation may lead to heart failure. Uncontrolled bleeding may occur from skin lesions during patient positioning and transport. Epistaxis may lead to aspiration of blood into trachea causing pulmonary edema. Intravenous access may be difficult. Sudden change in blood pressure may cause bleeding from AVMs anywhere in the body, most serious of which is from cerebral AVM. Gastric distension may occur from ingested blood and may cause reflux and aspiration during induction. Any instrumentation including laryngoscopy and intubation, nasogastric tube insertion, urinary catheterization should be carried out with utmost caution as bleeding may occur from undetected lesions.
Box 1: Perioperative management problems in HHT patient
|
In hemodynamically stable patients, posted for elective surgery, preoperative optimization of the anemic status is corrected with oral or parenteral iron and if necessary erythropoiesis-stimulating agent63. Preoperatively angiogenesis inhibitors or hormone therapy should be considered in selected patients to reduce perioperative bleeding. Careful history and physical examination may indicate any systemic involvement and standard radiological imaging with angiography may be performed to search for hemangiomas in brain, lung, gastrointestinal tract, nose and paranasal sinuses. In unstable patient presenting with severe bleeding focus should be directed to simultaneous resuscitation and hemostasis. Blood transfusion forms the mainstay of volume resuscitation in severely volume depleted patient. Epistaxis should be controlled with tight nasal packing immediately followed by cauterization of bleeding vessels and Septodermoplasty if required. Since bleeding does not result from a defect in coagulation cascade, but from the malformed vascular structures, platelet or plasma transfusions are of no use and reserved only to supplement the loss. Antifibrinolytics including tranexamic acid64,65 and aminocaproic acid66 have been used with success to control epistaxis. In addition to antifibrinolytic effects, tranexamic acid also stimulates the expression of ALK-1 and endoglin, as well as the activity of the ALK-1/endoglin pathway.67 Intraoperatively controlled hypotension should be achieved with nitroglycerine or inhaled anesthetics or alpha 2 agonists to reduce bleeding.
Conclusion
Patients with Osler-Weber-Rendu disease (OWRD) or Hereditary Hemorrhagic Telangiectasia (HHT) may present with uncontrolled bleeding. Resuscitation along with hemostasis forms the cornerstone of treatment. As the bleeding occurs from malformed vessels, standard coagulation tests will reveal no abnormality. Management strategies include blood transfusion, antifibrinolytics and surgical hemostasis. Anesthesia planning should include rapid sequence induction and controlled hypotension.
Conflict of interest: None declared by the authors
Authors’ Contribution:
SKK: Concept and writing
TG: Contributing author
References
- Peery WH. Clinical spectrum of hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease). Am J Med. 1987 May;82(5):989–97. [PubMed]
- Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995 Oct 5;333(14):918–24. [PubMed]
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000 Mar 6;91(1):66–7. [PubMed]
- Sharathkumar AA, Shapiro A. Hereditary haemorrhagic telangiectasia. Haemophilia. 2008 Nov;14(6):1269-80. doi: 10.1111/j.1365-2516.2008.01774.x. [PubMed] [Free full text]
- Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992 Aug:29(8):527-530. [PubMed] [Free full text]
- Goulart AP, Moro ET, Guasti VM, Colares RF. Anesthetic management of a patient with hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Case report. Rev Bras Anestesiol. 2009 Jan-Feb;59(1):74-8. [PubMed] [Free full text]
- Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999 Jan;245(1):31–9. [PubMed] [Free full text]
- Sutton H. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Med Mirror 1864; 1: 769–81.
- Rendu H. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muquez. Gazette des Hopitaux Civils et Militaires (Paris) 1896;135:1322–3.
- Osler W. On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes. Bull Johns Hopkins Hosp 1901; 12: 333–7.
- Weber F. Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet 1907; 2: 160–2.
- Hanes F. Multiple hereditary telangiectases causing hemorrhage (hereditary hemorrhagic telangiectasia). Bull Johns Hopkins Hosp 1909; 20: 63–73.
- Nanda S, Bhatt SP. Hereditary hemorrhagic telangiectasia: epistaxis and hemoptysis. CMAJ. 2009 Apr 14;180(8):838. doi: 10.1503/cmaj.081003. [PubMed] [Free full text]
- Shovlin CL, Letarte M. Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax. 1999 Aug;54(8):714-29. [Free full text]
- Ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004 May;29(5):265-73. [PubMed]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994 Aug 4;370(6488):341–7. [PubMed]
- Schmierer B, Hill CS. TGFbeta- SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007 Dec;8(12):970–82. [PubMed]
- Cirulli A, Liso A, D’Ovidio F, Mestice A, Pasculli G, Gallitelli M, et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003;110(1):29-32. [PubMed]
- Xu B, Wu YQ, Huey M, Arthur HM, Marchuk DA, Hashimoto T, et al. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab. 2004 Feb;24(2):237-44. [PubMed]
- McAllister KA1, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994 Dec;8(4):345-51. [PubMed]
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. Feb 2006;43(2):97-110. [PubMed] [Free full text]
- McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, et al. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat Genet. 1994 Feb;6(2):197–204. [PubMed]
- Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet. 1994 Feb;6(2):205–9. [PubMed]
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996 Jun;13(2):189-95. [PubMed]
- Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA, et al. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995 Aug;5(1):21-8. [PubMed] [Free full text]
- Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005 Jul;42(7):577-82. [PubMed] [Free full text]
- Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006 Oct 15;140(20):2155-62. [PubMed]
- Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004 Mar 13;363(9412):852-9. [PubMed]
- Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD, et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A. 2010 Feb;152A(2):333-9. doi: 10.1002/ajmg.a.33206. [PubMed]
- Iyer NK, Burke CA, Leach BH, Parambil JG. SMAD4 mutation and the combined syndrome of juvenile polyposis syndrome and hereditary haemorrhagic telangiectasia. Thorax. 2010 Aug;65(8):745-6. doi: 10.1136/thx.2009.129932. [PubMed] [Free full text]
- Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003 Dec;40(12):865-71. [PubMed] [Free full text]
- Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001 Aug 2;345(5):325-34. [PubMed] [Free full text]
- Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al.International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011 Feb;48(2):73-87. doi: 10.1136/jmg.2009.069013. Epub 2009 Jun 23. [PubMed]
- Schoen FJ. Cotran RS, Vinay K, Collins T. Robbins Pathologic Basis of Disease. 5th. WB Saunders; 1994:509
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989 Mar:32(3):291-297.[PubMed]
- Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003 Aug;40(8):585-90. [PubMed] [Free full text]
- Ingrosso M, Sabbà C, Pisani A, Principi M, Gallitelli M, Cirulli A, et al. Evidence of small-bowel involvement in hereditary hemorrhagic telangiectasia: a capsule-endoscopic study. Endoscopy. 2004 Dec;36(12):1074-9. [PubMed]
- Kjeldsen AD, Kjeldsen J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2000 Feb;95(2):415–8. [PubMed]
- Sabbà C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M, et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007 Jun;5(6):1149-57. [PubMed] [Free full text]
- Vase P, Holm M, Arendrup H. Pulmonary arteriovenous fistulas in hereditary hemorrhagic telangiectasia. Acta Med Scand. 1985;218(1):105–9. [PubMed]
- Haitjema T, Disch F, Overtoom TT, Westermann CJ, Lammers JW. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med. 1995 Nov;99(5):519–24. [PubMed]
- Fulbright RK, Chaloupka JC, Putman CM, Sze GK, Merriam MM, Lee GK, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neuroradiol. 1998 Mar;19(3):477-84. [PubMed]
- Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006 Jul;27(7):667–75.[PubMed]
- Matsubara S, Mandzia JL, ter Brugge K, Willinsky RA, Faughnan ME. Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. AJNR Am J Neuroradiol. 2000; 21:1016–20. [Free full text]
- Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabba C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia: CT findings. Abdom Imaging. 2004 Mar-Apr;29(2):211–20. [PubMed]
- Boyer H, Fernandes P, Duran O, Hunter D, Goding G. Office-based sclerotherapy for recurrent epistaxis due to hereditary hemorrhagic telangiectasia: a pilot study. Int Forum Allergy Rhinol. 2011 Jul-Aug;1(4):319-23. doi: 10.1002/alr.20053. Epub 2011 Apr 28. [PubMed]
- Mortuaire G, Boute O, Hatron PY, Chevalier D. Pilot study of submucosal radiofrequency for epistaxis in hereditary hemorrhagic telangiectasia. Rhinology. 2013 Dec;51(4):355-60. doi: 10.4193/Rhin13.027. [PubMed]
- Dheyauldeen S, Ostertun Geirdal A, Osnes T, Vartdal LS, Dollner R. Bevacizumab in hereditary hemorrhagic telangiectasia-associated epistaxis: effectiveness of an injection protocol based on the vascular anatomy of the nose. Laryngoscope. 2012 Jun;122(6):1210-4. doi: 10.1002/lary.23303. Epub 2012 May 7.[PubMed]
- Pau H, Carney AS, Murty GE. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): otorhinolaryngological manifestations. Clin Otolaryngol Allied Sci. 2001 Apr;26(2):93–8. [PubMed]
- Strother CM, Newton TH. Percutaneous embolisation to control epistaxis in Rendu-Osler-Weber disease. Arch Otolaryngol. 1976 Jan;102(1):58–60. [PubMed]
- Harries PG, Brockbank MJ, Shakespeare PG, Carruth JA. Treatment of hereditary haemorrhagic telangiectasia by the pulsed dye laser. J Laryngol Otol. Nov 1997;111(11):1038-41. [PubMed]
- Werner A, Bäumler W, Zietz S, Kühnel T, Hohenleutner U, Landthaler M. Hereditary haemorrhagic telangiectasia treated by pulsed neodymium:yttrium-aluminium-garnet (Nd:YAG) laser (1,064 nm). Lasers Med Sci. 2008 Oct;23(4):385-91. Epub 2007 Nov 13. [PubMed]
- Van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet. 1990 Apr 21;335(8695):953–5. [PubMed]
- Massoud OI, Youssef WI, Mullen KD. Resolution of hereditary hemorrhagic telangiectasia and anemia with prolonged alpha-interferon therapy for chronic hepatitis. C. J Clin Gastroenterol. 2004 Apr;38(4):377–9. [PubMed]
- Shovlin CL, Winstock AR, Peters AM, Jackson JE, Hughes JM. Medical complications of pregnancy in hereditary haemorrhagic telangiectasia. QJM. 1995 Dec;88(12):879–87. [PubMed]
- White RI Jr, Pollak JS, Wirth JA. Pulmonary arteriovenous malformations: diagnosis and transcatheter embolotherapy. J Vasc Interv Radiol. 1996 Nov-Dec;7(6):787– 804. [PubMed]
- Hadjipanayis CG, Levy EI, Niranjan A, Firlik AD, Kondziolka D, Flickinger JC, et al. Stereotactic radiosurgery for motor cortex region arteriovenous malformations. Neurosurgery. 2001 Jan;48(1):70–6. [PubMed]
- Levy EI, Niranjan A, Thompson TP, Scarrow AM, Kondziolka D, Flickinger JC, et al. Radiosurgery for childhood intracranial arteriovenous malformations. Neurosurgery. 2000;47:834–41. [PubMed]
- Dupuis-Girod S, Chesnais AL, Ginon I, Dumortier J, Saurin JC, Finet G, et al. Long-term outcome of patients with hereditary hemorrhagic telangiectasia and severe hepatic involvement after orthotopic liver transplantation: a single-center study. Liver Transpl. 2010 Mar;16(3):340-7. doi: 10.1002/lt.21990. [PubMed] [Free full text]
- Hillert C, Broering DC, Gundlach M, Knoefel WT, Izbicki JR, Rogiers X. Hepatic involvement in hereditary hemorrhagic telangiectasia: an unusual indication for liver transplantation. Liver Transpl. 2001 Mar;7(3):266–8. [PubMed] [Free full text]
- Lerut J, Orlando G, Adam R, Sabbà C, Pfitzmann R, Klempnauer J, et al. Liver transplantation for hereditary hemorrhagic telangiectasia: Report of the European liver transplant registry. Ann Surg. 2006 Dec;244(6):854-62; discussion 862-4. [PubMed] [Free full text]
- Mitchell A, Adams LA, Macquillan G, Tibballs J, Vanden Driesen R, Delriviere L. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl. 2008 Feb;14(2):210–3. doi: 10.1002/lt.21417. [PubMed] [Free full text]
- Cherif H, Karlsson T. Combination treatment with an erythropoiesis-stimulating agent and intravenous iron alleviates anaemia in patients with hereditary haemorrhagic telangiectasia. Ups J Med Sci. 2014 Nov;119(4):350-3. doi: 10.3109/03009734.2014.955619 [PubMed] [Free full text]
- Gaillard S, Dupuis-Girod S, Boutitie F, Rivière S, Morinière S, Hatron PY, et al. Tranexamic acid for epistaxis in hereditary hemorrhagic telangiectasia patients: a European cross-over controlled trial in a rare disease. J Thromb Haemost. 2014 Sep;12(9):1494-502. doi: 10.1111/jth.12654. Epub 2014 Jul 29. [PubMed] [Free full text]
- Morales-Angulo C1, Pérez del Molino A, Zarrabeitia R, Fernández A, Sanz-Rodríguez F, Botella LM. [Treatment of epistaxes in hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease) with tranexamic acid]. [Article in Spanish] Acta Otorrinolaringol Esp. 2007 Apr;58(4):129-32. [PubMed]
- Korzenik JR, Topazian MD, White R. Treatment of bleeding in hereditary hemorrhagic telangiectasia with aminocaproic acid. N Engl J Med. 1994 Nov 2;331(18):1236. [PubMed]
- Fernandez-L A, Garrido-Martin EM, Sanz-Rodriguez F, Ramirez JR, Morales-Angulo C, Zarrabeitia R, et al. Therapeutic action of tranexamic acid in hereditary haemorrhagic telangiectasia (HHT): regulation of ALK-1/endoglin pathway in endothelial cells. Thromb Haemost. 2007 Feb;97(2):254-62. [PubMed]
